 客服微v信:
客服微v信:
基于醫藥魔方網站行業快訊板塊、NextPharma、NextMed數據庫以及公開信息,2020年10月的《臨床研究月報》共篩選出19項值得關注的臨床研究,包括6項腫瘤研究,3項心血管研究、3項COVID-19研究和7項其他領域研究,供您參考。更多行業資訊,點擊文末閱讀原文,訪
基于醫藥魔方網站行業快訊板塊、NextPharma、NextMed數據庫以及公開信息,2020年10月的《臨床研究月報》共篩選出19項值得關注的臨床研究,包括6項腫瘤研究,3項心血管研究、3項COVID-19研究和7項其他領域研究,供您參考。更多行業資訊,點擊文末閱讀原文,訪問醫藥魔方官方網站。
腫瘤領域
1. Opdivo+Yervoy
10月2日,百時美施貴寶(BMS)公布了Opdivo與Yervoy免疫組合療法治療IIIB/C/D期或IV期黑色素瘤的III期臨床研究(CheckMate-915)的共同主要終點結果。結果顯示,在所有患者(意向性治療,ITT)中,與Opdivo單藥治療相比,O+Y組合在無復發生存期(RFS)的共同主要終點上,沒有達到統計學意義的顯著改善。
CheckMate -915是一項隨機、雙盲、安慰劑對照的III期研究,評估了已完全手術切除的IIIB/C/D期或IV期(無疾病證據)黑色素瘤患者使用Opdivo聯合Yervoy與Opdivo單藥療法(已批準的標準治療)的治療效果。入組研究的患者除了接受黑素瘤病灶手術和/或中樞神經系統病灶的神經外科切除術后的輔助放射治療外,之前沒有接受過黑素瘤的抗癌治療。該研究中,共有1934例患者進行了隨機分配,接受O+Y組合療法(Opdivo 240mg,每兩周靜脈輸注一次;Yervoy 1mg/kg,每六周靜脈輸注一次)或Opdivo單藥治療(480mg,每4周靜脈輸注一次),治療時間為一年。此前,2019年11月,BMS曾宣布,在腫瘤已切除且表達PD-L1<1%的高危黑色素瘤患者中,與Opdivo單藥治療相比,O+Y組合在無復發生存期(RFS)的共同主要終點沒有達到統計學意義的顯著改善。
雖然試驗取得陰性結果,但是CheckMate-915結果強化了Opdivo單藥在輔助治療中作為標準護理療法的既定益處。BMS將完成對CheckMate-915數據的全面評估,并與研究人員在即將召開的醫學會議上分享結果。
2. Sotorasib(AMG 510)
10月5日,安進宣布其KRAS G12C抑制劑sotorasib(AMG 510)治療126例晚期非小細胞肺癌(NSCLC)患者的II期CodeBreaK 100臨床試驗頂線結果呈陽性,這些患者此前接受過免疫治療和/或化療。
研究結果表明,Sotorasib的客觀應答率與先前報道的 I 期臨床(960mg/日劑量組治療晚期NSCLC)數據一致,到達主要終點。其他包括應答持續時間在內的療效指標也令人鼓舞,數據截止時有超過一半的應答患者仍在接受治療且持續應答。安全性與耐受性與既往研究結果一致。這項潛在性II期注冊性臨床研究數據將在2021年1月舉辦的世界肺癌大會中進行公布。
3. Opdivo+化療
10月7日,BMS宣布,在可切除的NSCLC患者中進行Opdivo聯合化療的III期臨床研究(CheckMate-816)達到病理完全緩解(pCR)的主要研究終點。研究結果顯示,在術前接受納武利尤單抗聯合化療的患者中,手術切除標本未發現癌細胞的患者人數顯著多于單用化療的患者。CheckMate-816成為首個,也是目前唯一一個證實免疫檢查點抑制劑聯合化療作為新輔助治療能夠為非轉移性非小細胞肺癌患者帶來獲益的III期臨床研究。
Checkmate-816是一項隨機、開放標簽、多中心的III期臨床研究,旨在評估與單用化療相比,納武利尤單抗聯合化療用于可切除非小細胞肺癌患者新輔助治療的療效。在主要分析中,358例患者在術前隨機接受納武利尤單抗(360 mg)聯合基于組織學分型的含鉑雙藥化療(每3周一次,最多3個周期),或者單用含鉑雙藥化療(每3周一次,最多3個周期)。主要研究終點是病理完全緩解(pCR)和無事件生存期(EFS),關鍵次要終點包括總生存期(OS)、主要病理緩解(MPR),以及至死亡或遠處轉移的時間。
BMS將完成對CheckMate-816研究現有數據的全面評估,與研究者共同在即將舉行的醫學會議上公布研究結果,并將與衛生監管部門討論潛在的注冊路徑。CheckMate-816研究目前仍在進行中,以評估另一個主要研究終點無事件生存期(EFS)以及關鍵次要終點,目前此部分數據對公司依然為盲態。
迄今為止,歐狄沃作為新輔助或輔助治療已在四種腫瘤類型中顯示出了療效改善,包括肺癌、膀胱癌、食管癌/胃食管連接部癌和黑色素瘤。
4. Ibrance(palbociclib)
10月9日,輝瑞宣布其CDK4/6抑制劑Ibrance(palbociclib)在治療HR+、HER2-女性早期乳腺癌患者的III期PENELOPE-B研究中未到達無浸潤性疾病生存期 (iDFS)的主要終點,這些患者在完成新輔助化療后仍有殘留浸潤性疾病。
PENELOPE-B研究是一項隨機、雙盲、安慰劑對照的III期臨床試驗,共納入1250例患者,旨在比較Ibrance聯合內分泌標準治療較安慰劑聯合內分泌標準治療的療效和安全性。這些患者臨床病理分期-雌激素/分級(CPS-EG)評分為3分或更高 (如果手術時有淋巴結轉移,則為2分)。研究未觀察到新的安全性信號。詳細試驗數據將在之后進行的醫學會議上公布。
這也是Ibrance(palbociclib)今年5月在聯合內分泌療法治療HR+/HER2-的III期臨床研究PALLAS遭遇失敗之后,在早期乳腺癌臨床研究的又一次失利。而此次PENELOPE-B的臨床試驗在早期乳腺癌治療中未達到主要終點,亦沒有觀察到意外的安全信號。輝瑞方面表示,盡管對這一結果感到失望,仍然希望繼續與研究合作伙伴合作,以了解這些數據如何為早期乳腺癌下一代CDK抑制劑的開發提供信息。
5. SGI-110
10月14日,Astex和Otsuka公布了Guadecitabine(SGI-110)的III期試驗ASTRAL-2(NCT02920008)和ASTRAL-3(NCT02907359)頂線數據,用于既往接受過治療的成人患者治療急性髓性白血病(AML)和骨髓增生異常綜合征或慢性粒單核細胞白血病(MDS/CMML)。研究結果顯示,SGI-110對比醫生選擇的替代療法未顯著改善OS,兩項試驗均未達到主要終點。目前正在評估研究的預期亞組和次要終點,完整數據將在科學會議上呈現。
Guadecitabine是下一代DNA甲基化劑,經過合理設計,可抵抗胞苷脫氨酶降解,從而延長腫瘤細胞對活性代謝產物地西他濱的暴露時間,通過地西他濱的作用抑制DNA甲基轉移酶(DNMT),具有逆轉異常DNA甲基化的潛能。DNA甲基化是許多癌細胞的表觀遺傳學特征,并可能恢復沉默的抑癌基因和腫瘤相關抗原的表達,通過這種沉默基因的重新表達,Guadecitabine可能具有使腫瘤細胞對其他抗癌藥(包括免疫治療藥物)的敏感性,以及使先前對化學治療有抗性的癌細胞致敏的潛力。
6. Adagrasib(MRTX849)
10月25日,Mirati Therapeutics在第32屆國際分子靶標與癌癥治療學研討會(EORTC-NCI-AACR)上發表KRAS G12C選擇性抑制劑adagrasib(MRTX849)在攜帶KRAS G12C突變的晚期/轉移性非小細胞肺癌和結直腸癌中的最新臨床數據。
結果顯示,在晚期/轉移性非小細胞肺癌中,45%的患者對adagrasib(MRTX849)治療具有客觀反應,70%的患者應答者對腫瘤的最佳應答大于40%,患者的疾病控制率為96%。在可評估的CRC患者中,17%(3/18)有確定的客觀反應,并且3例患者中有2例仍在接受治療。94%(17/18)的患者中觀察到疾病控制,并且仍有12例患者仍在接受治療,55%(10/18)的患者治療時間超過4個月。更多詳細內容可參閱此前報道《KRAS抑制劑對比:MRTX849比AMG510更好嗎?》。

心血管領域
1. Omecamtiv mecarbil
10月08日,安進與其合作伙伴Cytokinetics和施維雅(Servier)共同宣布了omecamtiv mecarbil在射血分數降低(HFrEF)的心力衰竭患者中進行的關鍵III期臨床研究(GALACTIC-HF)頂線結果。結果顯示,omecamtiv mecarbil達到了與安慰劑相比,使心血管(CV)死亡或心衰事件(心衰住院或其他心衰緊急治療)在統計學上顯著減少(HR=0.92;95%CI:0.86,0.99;p=0.0252)的主要復合療效終點,沒有觀察到心血管死亡的次要終點降低。不良事件(包括重大缺血性心臟事件)在各治療組之間未顯示差異。GALACTIC-HF的結果將在2020年11月13日美國心臟協會(AHA)的科學會議上進行介紹。
GALACTIC-HF(通過改善心力衰竭的收縮力降低不良心臟結局的全球方案)是迄今為止在心衰治療領域開展的最大規模III期全球性心血管結局研究之一,在35個國家入組了8256例HFrEF患者,這些患者紐約心臟協會(NYHA)評級為II-IV級、左室射血分數(LVEF)≤35%、利鈉肽升高,且在入組研究時因心衰住院,或在篩查前一年內因心衰住院或進入急診室。研究的目的是評估在標準護理中加入omecamtiv mecarbil治療是否能降低HFrEF患者發生心衰事件(心衰住院治療和其他心衰緊急治療)和心血管(CV)死亡的風險。將患者隨機分為口服安慰劑或25 mg omecamtivmecarbil起始劑量,每天兩次(維持劑量分別為50 mg,37.5mg或25 mg,每天兩次)。
Omecamtiv mecarbil是一種研究性的選擇性新型心肌肌球蛋白激活劑,是首個新型肌營養劑。臨床前研究表明,心肌肌球蛋白激活劑可在不影響心肌細胞胞內鈣濃度或心肌耗氧量的情況下增加心肌收縮力。心肌肌球蛋白是心肌細胞中的細胞骨架運動蛋白,直接負責將化學能轉化為導致心肌收縮的機械力。
2. Jardiance(empagliflozin)
10月23日,勃林格殷格翰和禮來公司發布公告,在EMPEROR-Reduced III期臨床試驗新的探索性亞組分析中表明,恩格列凈可降低伴或不伴糖尿病、射血分數降低的成年人心力衰竭患者的心血管和腎臟不良事件的風險,無論其基礎性慢性腎臟疾病情況如何。
如先前報道,EMPEROR-Reduced顯示Jardiance將患有或未患有糖尿病的心力衰竭伴有射血分數降低的成年人的心血管死亡或因心力衰竭住院的綜合終點的相對風險降低了25%,將因心力衰竭而首次住院和反復住院的相對風險降低了30%,并減緩了eGFR(衡量腎功能下降的指標)的下降。另一項探索性分析表明,Jardiance將復合腎終點(包括終末期腎臟疾病和腎功能嚴重喪失)的相對風險降低了50%。在這項新分析中的所有終點中,在基線時有或沒有慢性腎臟病的患者(包括嚴重腎功能不全的患者)的亞組中均觀察到了這些益處。在參加EMPEROR-Reduced試驗的所有患者隊列中,安全性與Jardiance已知的安全性相似。
3. Forxiga(dapagliflozin)
10月,國家藥品監督管理局(NMPA)批準更新阿斯利康安達唐(達格列凈,dapagliflozin)中國說明書,納入了DECLARE-TIMI 58 III期臨床研究結果中的部分數據。
DECLARE-TIMI 58研究是一項由阿斯利康申辦的隨機、雙盲、安慰劑對照、多中心的 III期臨床研究,旨在評估達格列凈與安慰劑相比,對具有心血管事件風險(包括多重心血管危險因素或已確診的心血管疾病)的2型糖尿病(T2D)成年患者的心血管結局,同時也評估了關鍵腎臟次要終點。DECLARE-TIMI 58研究納入了來自33個國家、882個中心的17,000多例患者,由TIMI研究組(馬薩諸塞州,波士頓)獨立運行,并與哈達薩希伯來大學醫學中心(以色列,耶路撒冷)合作。
DECLARE-TIMI 58研究顯示:與安慰劑相比,達格列凈能夠有效降低已確診心血管疾病或多重心血管危險因素的成人2型糖尿病患者因心衰住院(hHF)或心血管死亡的風險。該研究所示安全性與達格列凈已知安全性一致。
COVID-19
1. LY-CoV555+LY-CoV016
10月7日,禮來宣布了其抗SARS-CoV-2病毒中和抗體開發項目的最新進展,包括兩款中和抗體 LY-CoV555與LY-CoV016(從君實引進的JS016)的聯合療法用于新確診輕至中度COVID-19患者的BLAZE-1研究最新期中數據。
BLAZE-1研究(NCT04427501)為一項隨機、雙盲、安慰劑對照、II期研究,主要評估LY-CoV555和LY-CoV016相比安慰劑用于治療伴有癥狀COVID-19門診患者的療效及安全性差異,計劃入選800例癥狀輕至中度且藥物輸注前3日內SARS-CoV-2樣本檢測為陽性的COVID-19患者。
該研究的單藥治療隊列評估LY-CoV555三個劑量 (700,2800,7000mg)的治療效果和安全性,聯合用藥組評估LY-CoV555(2800mg)+LY-CoV016(2800mg)的治療效果和安全性。安慰劑組患者在完成各隊列研究后允許進入全部受試藥組交叉用藥。研究主要終點是第11天SARS-CoV-2 病毒載量較基線的變化。其他終點包括因COVID-19住院的患者比例,29天內死亡率、安全性等。
截至期中分析時,聯合療法組入組112人,安慰劑對照組入組156人。結果顯示,LY-CoV555+ LY-CoV016聯合療法顯著降低了第11天的病毒載量(p=0.011),達到了該研究的主要終點。聯合療法還降低了第3天(p=0.016)及第7天(p<0.001)(感染過程中較早的時間點,通常能觀察到更高的病毒載量)的病毒水平。該聯合療法亦達到預設的臨床終點,包括自第1至11天的癥狀總體評分相較于基線的時間加權平均變化。探索性分析結果顯示,聯合療法組第7天病毒載量依然較高的患者比例低于安慰劑對照組(3% vs 20.8%,p<0.0001)。聯合療法組COVID-19相關住院和急診治療的發生率低于安慰劑對照組(0.9% vs 5.8%),風險降低84.5%。
2. Merimepodib+ Remdesivir
10月26日,BioSig Technologies及其控股子公司ViralClearPharmaceuticals宣布停止Merimepodib聯用瑞德西韋(Remdesivir)在晚期冠狀病毒病(COVID-19)中的臨床開發。
該決定是基于一項隨機、雙盲、安慰劑對照的II期研究,評價口服Merimepodib與靜脈注射瑞德西韋聯用治療成人冠狀病毒性疾病患者(COVID- 19)的療效。實施方案修訂后,將試驗的規模從40位住院的COVID-19患者擴大到80位,并且僅限于重癥患者入組(NIAID 3級,需要高流量、高濃度的氧氣)。安全監督委員會(SMC)在對數據進行最新審查時,已有44例患者入組,其中42例接受了研究藥物(Merimepodib或安慰劑)。
數據的最新審查結果表明,所有22位4級患者均已出院,并且在37天的隨訪期內未復發。但是,入組時為NIAID 3級患者(n=20)的患者結局明顯不同,具體而言,無盲SMC監測到這些NIAID 3級患者的生存率在安慰劑和merimepodib之間的不平衡(imbalance in survivalrates),因此該試驗不太可能達到其主要安全性終點。因此,該公司選擇停止注冊該臨床試驗。將按照方案對患者進行安全監控;但是,將不會進行進一步的藥物研究治療。
3. REGN-COV2
10月28日,再生元(Regeneron)宣布,其新冠中和抗體雞尾酒療法REGN-COV2在一項正在進行的II/III期臨床試驗中達到主要和關鍵性次要終點。REGN-COV2顯著降低患者的病毒載量和接受進一步醫療護理的需求(包括住院、急診室就診、緊急護理和/或醫生辦公室/遠程醫療訪視)。
這項隨機、雙盲試驗正在評估將REGN-COV2加入到常規護理標準中與將安慰劑加護理標準中相比的效果。先前報道了對前275名患者的描述性分析,此次報告的數據涉及另外524名患者。
結果顯示,在病毒學結果方面,接受治療7天后,新增患者(n=524)的平均病毒載量與對照組相比降低0.36個log10(p=0.0003,1個log10為10倍)。在基線病毒載量更高的患者中(定義為病毒載量超過1X107拷貝/毫升),REGN-COV2與安慰劑組相比將病毒載量降低0.68個log10(p<0.0001)。在接受治療后第5天,REGN-COV2與對照組相比,將病毒載量降低1.08個log10,意味著這些患者的病毒載量與對照組相比降低10倍以上。在關鍵的臨床終點上,在第29天使用REGN-COV2治療可使COVID-19相關的醫療就診次數減少了57%(2.8%聯合劑量組;6.5%安慰劑;p = 0.024)。在具有一種或多種危險因素(包括50歲以上;體重指數大于30;心血管、代謝、肺、肝或腎疾病;包括糖尿病或免疫功能低下)的患者中,使用REGN-COV2治療可將COVID-19相關的醫療就診次數減少72%。
結果還顯示,高劑量(8克)和低劑量(2.4克)之間的REGN-COV2病毒學或臨床療效無明顯差異。基于這一發現,鑒于目前REGN-COV2的供應有限,Regeneron正在審查正在進行的門診臨床試驗中劑量的潛在變化,并計劃尋求緊急授權使用。
然而,10月30日,再生元再次發布公告,由于存在潛在的安全隱患,已暫停了用于治療重癥新冠患者抗體藥物的臨床研究。再生元公司表示,針對新冠病毒的抗體雞尾酒療法試驗的獨立數據監測委員會(IDMC)建議“修改”患者試驗,因此部分患者的招募計劃被擱置。獨立數據監測委員會(IDMC)在一份聲明中表示,建議在進一步的數據分析前,暫停進一步招募需要大量吸氧或呼吸機的患者,但是可以繼續招募不需要吸氧或只需要少量吸氧的輕癥患者,并繼續在不加修改的情況下進行門診試驗。
其他
1. PXL770
10月01日,POXEL SA發布公告稱,PXL770用于NASH治療的IIa期臨床試驗達到了其主要療效終點,患者在接受12周治療時,肝脂肪相對減少方面具有統計學上的顯著改善,在2型糖尿病患者中效果更加顯著。在接受PXL770治療的患者中,主要的次要措施包括肝酶-丙氨酸轉氨酶(ALT)和血紅蛋白A1c(HbA1c)的統計學改善,并且PXL770安全且耐受性良好。
這也是直接AMPK激活劑的首次人類臨床評估,結果支持包括主要高風險亞組(2型糖尿病患者)在內的NASH治療潛力,以及AMPK激活劑在其他慢性和罕見代謝疾病中的應用。
2. Zeposia(ozanimod)
10月10日,BMS宣布了True North研究的詳細結果。True North是一項關鍵、安慰劑對照的III期試驗,評估口服Zeposia(ozanimod)作為中度至重度潰瘍性結腸炎(UC)成年患者的誘導和維持療法。True North達到了兩個主要終點,在第10周誘導期(18.4% vs 6.0%;p<0.0001)和52周維持期(37.0%vs 18.5%,p <0.0001),與安慰劑相比,具有較高的統計學意義和臨床意義。該研究還達到了關鍵的次要終點,包括臨床反應、內窺鏡改善和第10周誘導時的黏膜愈合以及第52周維持情況。與安慰劑相比,Zeposia治療的患者在第10周達到臨床反應的比例明顯更高(47.8% vs 25.9%;p<0.0001)和第52周時(60.0% vs 41.0%;p <0.0001)獲得臨床療效的患者明顯多于安慰劑組,且各子分析結果一致。觀察到的總體安全性與Zeposia和中度至重度UC患者的已知安全性一致。
3. Filgotinib
10月12日,吉利德和GalapagosNV更新了Filgotinib(口服、每天一次的JAK1抑制劑)用于治療中度至重度活動性潰瘍性結腸炎(UC)的最新數據,證明了其持續的療效和安全性。來自隨機、雙盲、安慰劑對照的IIb/III期SELECTION試驗數據表明,與安慰劑相比,200 mg filgotinib治療的患者在第10周達到臨床緩解并在58周之前保持緩解的比例顯著更高。此外,顯著更多的患者實現了六個月無皮質類固醇的緩解。
基于該數據,11月02日,EMA批準Filgotinib的新適應癥申請,用于治療對常規療法或生物制劑的反應不足、反應遲緩或不耐受的中度至重度活動性潰瘍性結腸炎(UC)的成年患者。
4. Dupixent
10月13日,賽諾菲宣布其Dupixent(dupilumab)在治療6-11歲兒童中重度哮喘的關鍵性III期臨床試驗中到達主要終點和關鍵的次要終點。Dupixent也是目前唯一一個在隨機III期研究中證明能改善兒童肺功能的生物制劑。
這項代號為LIBERTY ASTHMAVOYAGE的隨機、雙盲、安慰劑對照的III期臨床試驗,旨在評估Dupixent聯合標準療法對于兒童中重度哮喘患者的療效和全性。主要終點為評估2個預先指定人群的重度哮喘發作年化率:嗜酸性粒細胞(EOS)≥300 cells/μl和具有2型炎癥標志物(FeNO ≥20 ppbor EOS ≥150 cells/μl)患者。結果顯示,接受Dupixent治療的患者較安慰劑組重度哮喘年化率分別降低了65%和59%。與基線期相比,Dupixent治療組患者肺功能分別改善了10.15和10.53個百分點,而安慰劑組為4.83和5.32個百分點。安全性數據與既往結果一致。
5. AMX0035
10月16日,Amylyx公布了AMX0035治療肌萎縮側索硬化癥的II/III期試驗(CENTAUR)數據。研究結果顯示,隨訪期間,AMX0035相比安慰劑降低死亡風險達44%(HR,0.56;95%CI,0.34-0.92;P=0.023)。此前披露數據顯示,AMX0035 vs. 安慰劑的ALSFRS-R得分平均變化率為-1.24 vs -1.66。這是首個同時顯示長期生存獲益和功能改善的治療ALS試驗。
AMX0035是一種研究性神經保護療法,旨在通過靶向線粒體和內質網相關的細胞死亡途徑來減少運動神經元的死亡和功能障礙。這些途徑代表了與目前批準的藥物利魯唑(Riluzole)和依達拉奉(Edaravone)不同。大多數CENTAUR參與者(77%)在試驗進入期間和/或之前接受了批準的ALS治療(利魯唑、依達拉奉或兩者兼有),并被允許在OLE期間繼續使用這些藥物。生存分析比較了最初隨機分配給AMX0035的參與者和最初隨機分配給安慰劑的參與者之間的死亡時間(全因死亡率),在隨機分組后的35個月中,在隨訪過程中,死亡風險降低了44%,AMX0035的生存獲益與利魯唑和/或依達拉奉或兩者并用的基線使用無關。
6. SHR0302
10月26日,瑞石生物醫藥(瑞石生物)宣布,其在研的國內首創、具有自主知識產權的高選擇性JAK1抑制劑SHR0302用于治療特應性皮炎的II期臨床研究(QUARTZ2)獲得成功,達到試驗主要及次要終點。
這項隨機、雙盲、安慰劑對照、多中心臨床研究在中國23個研究中心進行。納入研究的105名成人受試者均為既往疾病控制不佳的患者,被隨機分配每日口服8mg或4mg的SHR0302片或安慰劑,療程為12周。主要終點為第12周時達到研究者總體評分(IGA)應答的受試者百分比。研究結果顯示,兩種治療劑量均明顯改善了患者的臨床癥狀,并提高了生活質量。患者在接受了8mg或4mg SHR0302片單一療法后,皮膚病灶清除顯著有效。12周時,接受8mg治療的患者中54.3%獲得IGA應答,接受4mg治療的患者中25.7%達到IGA應答(8mg vs 安慰劑,p<0.001;4mg vs 安慰劑,p=0.022)。安全性方面,SHR0302總體耐受性良好。試驗中沒有死亡或危及生命的病例報告,沒有嚴重感染的病例報告,也沒有惡性腫瘤或血栓形成的情況。
7. Upadacitinib
10月29日,在第29屆歐洲皮膚病和性病學會(EADV)虛擬大會上,艾伯維(Abbvie)公布了來自Measure Up 1和2的upadacitinib單藥療法研究(RINVOQ)的新分析數據,與安慰劑相比,使用upadacitinib(15 mg或30 mg;每天一次)治療的皮炎患者在皮膚清除率、瘙癢和生活質量的其他衡量指標有所改善,沒有發現新的安全風險。
數據來自III期Measure Up 1和Measure Up 2研究,結果支持最近向美國食品藥品監督管理局和歐洲藥品管理局提交的申請,尋求批準upadacitinib在成人和青少年中度至重度特應性皮炎患者中的使用批準。在MeasureUp 1和2兩項研究中,在第16周,較接受安慰劑治療的患者相比,接受兩種劑量的upadacitinib治療的患者的濕疹面積嚴重性指數(EASI 90)至少改善了90%以上(p<0.001);此外,對于兩種劑量的upadacitinib,在第4周時達到臨床意義(NRS≥4.1)的瘙癢減輕患者比例顯著高于安慰劑,并維持到第16周。今年早些時候,AbbVie公布了MeasureUp 1和Measure 2研究的主要數據,顯示upadacitinib(15 mg或30 mg)達到了共同主要終點和所有次要終點(p <0.001)。
本文源自頭條號:醫藥魔方,作者:。版權歸原作者所有。
加強版PI3K抑制劑來了!高效抑制小鼠肺纖維化,提高生存率
特發性肺纖維化(idiopathic pulmonary fibrosis,IPF)是一種致命的肺部疾病,好發于中老年人。由于膠原蛋白過度沉積,肺實質逐漸變硬變厚,導致肺部的氣體交換功能逐漸失效,患者的預期壽命僅為3-5年。雖然FDA已經批準了兩種治療特發性肺纖維化的藥物(吡非...
本文來源:醫藥魔方Plus 作者:小編 免責聲明:該文章版權歸原作者所有,僅代表作者觀點,轉載目的在于傳遞更多信息,并不代表“醫藥行”認同其觀點和對其真實性負責。如涉及作品內容、版權和其他問題,請在30日內與我們聯系

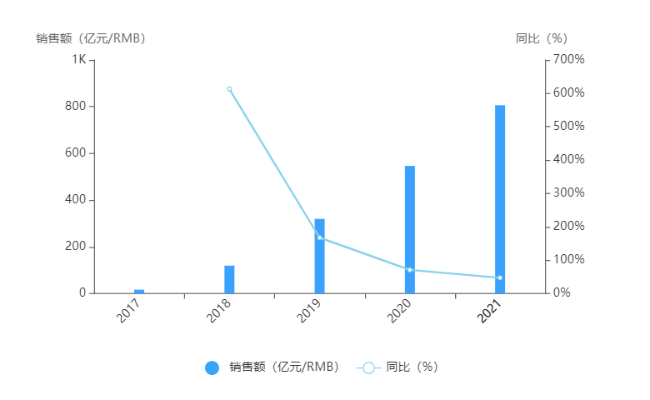
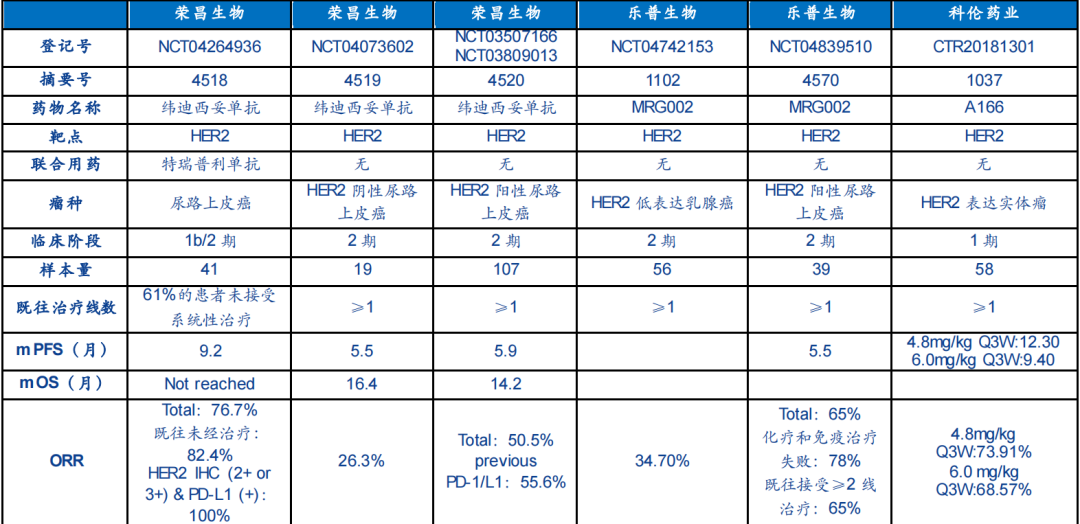

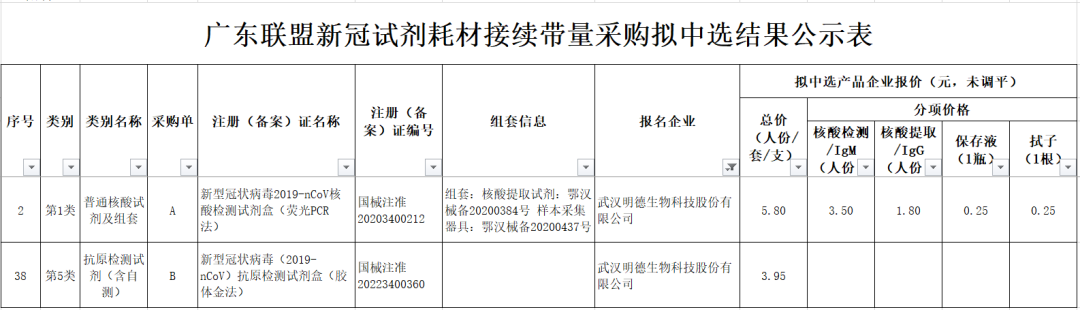

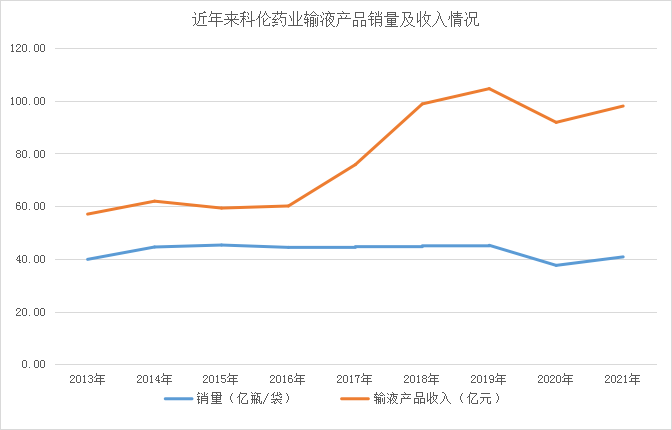



 京公網安備 11010802031568號
京公網安備 11010802031568號

